

What is a FDA medical device? Overview of Medical Device Classification
Medical devices are among the products that directly impact the health and well-being of users. Therefore, ensuring the quality and safety of medical devices is of utmost importance.In the United States, the Food and Drug Administration (FDA) is the authorized agency responsible for managing and overseeing the quality of medical devices. The FDA has established strict regulations for medical devices to ensure that they are manufactured and distributed in the U.S. market with the highest standards of quality and safety. Let’s explore FDA medical devices and the classification of medical devices according to the FDA in the article below with GOL!
1. What is a FDA medical device?
FDA medical devices are products designed, manufactured, or used for diagnosis, prevention, treatment, or alleviation of diseases; for birth control; to regulate the structure or function of the human body; or to support, replace, or modify human body parts.
FDA stands for the Food and Drug Administration of the United States, a federal agency responsible for ensuring the safety of food, drugs, and medical devices. The FDA has established strict regulations for medical devices to ensure that they are manufactured and circulated in the U.S. market with the highest standards of quality and safety.

2. FDA medical device classification
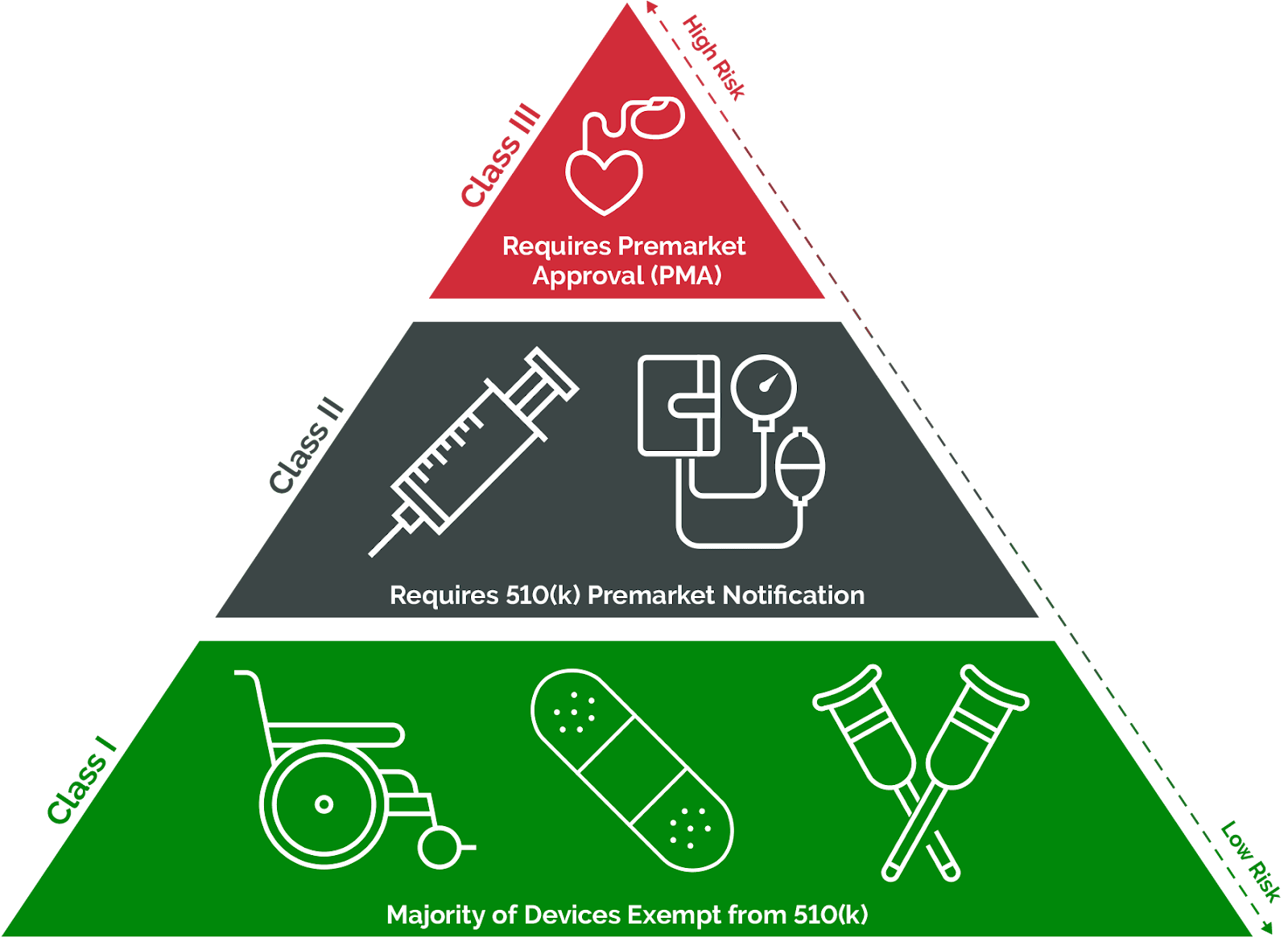
FDA classifies medical devices into three categories based on the potential risk they pose to users:
- Class I: Devices with low risk, such as thermometers, medical gloves, and safety glasses.
- Class II: Devices with moderate risk, including syringes, blood pressure monitors, and heart rate monitors.
- Class III: Devices with high risk, such as pacemakers, ventilators, and implantable devices.
FDA Class I and II medical devices must meet FDA requirements for design, manufacturing, and quality testing. FDA Class III medical devices undergo clinical evaluation to demonstrate effectiveness and safety before being brought to the market.
Compliance with FDA regulations for medical devices is crucial to ensure user safety. Manufacturers of FDA-regulated medical devices must meet FDA requirements to market their products in the United States.
Read more: What is the frequency of medical device audits by the FDA?
2.1 FDA Class I medical device
The FDA defines Class I devices as:
“Not intended for use in supporting or sustaining life or of substantial importance in preventing impairment to human health, and they may not present a potential unreasonable risk of illness or injury.”
These devices constitute the most prevalent class regulated by the FDA, accounting for 47% of approved devices in the market.
Class I devices involve minimal patient contact and have a negligible impact on overall patient health. Typically, they avoid contact with a patient’s internal organs, central nervous system, or cardiovascular system.
These devices are subject to the least stringent regulatory requirements.
Examples of Class I medical devices include:
- Electric toothbrush
- Tongue depressor
- Oxygen mask
- Reusable surgical scalpel
- Bandages
- Hospital beds
- Non-electric wheelchair
Bringing Class I medical devices to market is, as expected, the quickest and easiest, given their low risk to patients and infrequent criticality in life-sustaining care. The majority of Class I devices are exempt from FDA requirements for Premarket Notification (510(k)) and Premarket Approval (PMA).
However, Class I devices are not exempt from FDA General Controls, a set of regulations applicable to Class I, II, and III medical devices. General Controls cover aspects like adulteration, misbranding, device registration, records, and good manufacturing practices.
Manufacturers of Class I medical devices still need to establish a quality management system and adhere to standards to ensure a high-quality product.

2.2 What is FDA Class II medical device
Class II medical devices are more complex than Class I devices and pose a higher level of risk due to their potential for continuous contact with patients. This may include devices that come into contact with the circulatory system or internal organs and diagnostic tools.
The FDA defines Class II devices as:
“devices for which general controls are insufficient to provide reasonable assurance of the safety and effectiveness of the device.”
Examples of Class II medical devices:
- Catheters
- Blood pressure cuffs
- Pregnancy test kits
- Syringes
- Blood transfusion kits
- Contact lenses
- Surgical gloves
- Absorbable sutures
The process of bringing Class II medical devices to the market requires various controls depending on the type of device, but according to the FDA, it may include:
- Device performance
- Post-market surveillance
- Patient registries
- Special labeling requirements
- Premarket data requirements
- Guidelines
Class II devices are subject to the same General Controls mentioned above, as well as Special Controls. These regulations vary depending on the device and may encompass specific labeling requirements, patient registries, and performance standards.
Most notably, the majority of Class II devices enter the market through the premarket notification 510(k) process. The 510(k) is a comprehensive application submitted to the FDA, demonstrating the device’s safety and effectiveness by establishing its equivalence to another device already on the market.
This involves establishing ‘substantial equivalence’ to the existing marketed device, referred to as the ‘predicate’ in FDA terminology. This doesn’t necessitate identical devices, but there should be significant similarities in terms of use, design, materials, labeling, standards, and other characteristics.
2.3 FDA Class III medical device
The FDA defines Class III devices as products that:
“usually sustain or support life, are implanted or present a potential unreasonable risk of illness or injury.“
Only 10% of devices regulated by the United States Food and Drug Administration (FDA) fall under Class III. This classification is typically applied to permanent implants, smart medical devices, and life-support systems.
Class III medical device examples
- Breast implants
- Pacemakers
- Defibrillators
- High-frequency ventilators
- Cochlear implants
- Fetal blood sampling monitors
- Implanted prosthetics
Class III devices must comply with all General Controls and the FDA’s Premarket Approval (PMA) process. The FDA states that ‘only general and special controls alone are not enough to ensure the safety and effectiveness of Class III devices.’
Consistent with this, PMA is the most stringent type of marketing application required by the FDA for medical devices.
As you can see in our decision tree above, some FDA Class III devices may qualify for the 510(k) pathway if you can find a suitable predicate device marketed before the Medical Device Amendments of 1976. However, over time, this pathway has become more challenging and less common, as the FDA is less inclined to use devices older than a decade as predicates. Therefore, PMA is the most common choice for your Class III device.
The PMA process and premarket evaluation require a meticulous study of your Class III medical device to demonstrate its safety and effectiveness through the development of a risk/benefit profile based on data.
The PMA process typically involves clinical trials and a substantial amount of time and resources to gather sufficient data. The FDA will also conduct a rigorous assessment of your quality system during this period.
Read more: What is ISO 13485? Detailed explanation of the standard
3. A reputable company providing FDA medical device registration services
Due to the challenging and complex nature of the FDA medical device registration process, most businesses turn to FDA registration service companies for assistance. These companies help businesses prepare registration documentation, address inquiries regarding FDA regulations, and monitor the FDA filing process.GOL is a specialized company providing consulting and support services to businesses in FDA registration. With a team of experienced and well-trained experts in FDA regulations, GOL is ready to assist businesses at every stage of the FDA medical device registration process. Contact us here now for dedicated support!
Related articles:
