
Explaining the Differences of the 3 FDA Medical Device Classes
Success in conquering the U.S. medical device market requires a clear understanding of the FDA classification system. In this article, GOL will provide you with knowledge about the three FDA medical device classes: Class I, Class II, and Class III, along with the regulatory requirements and market approval pathways for each type.
What are FDA medical device classes?
The FDA diligently oversees the safety and efficacy of medical devices in the U.S. by categorizing them into different classes based on their risk profiles. This classification system ensures that appropriate levels of regulatory control are applied to each device to safeguard patients and users.
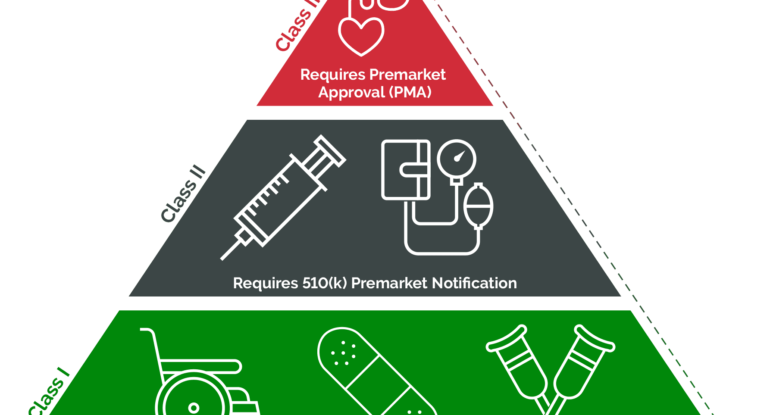
Class I devices, such as bandages and tongue depressors, are considered low risk and are subject to general controls, while Class II devices, including syringes and infusion pumps, require greater oversight, typically necessitating a 510(k) clearance. At the top of the hierarchy are Class III devices, like pacemakers and implantable defibrillators, which undergo the most stringent scrutiny, often requiring premarket approval (PMA) from the FDA to demonstrate their safety and effectiveness. This tiered approach helps maintain high standards of safety and efficacy across the spectrum of medical devices available in the market.
The differences between FDA medical device classes
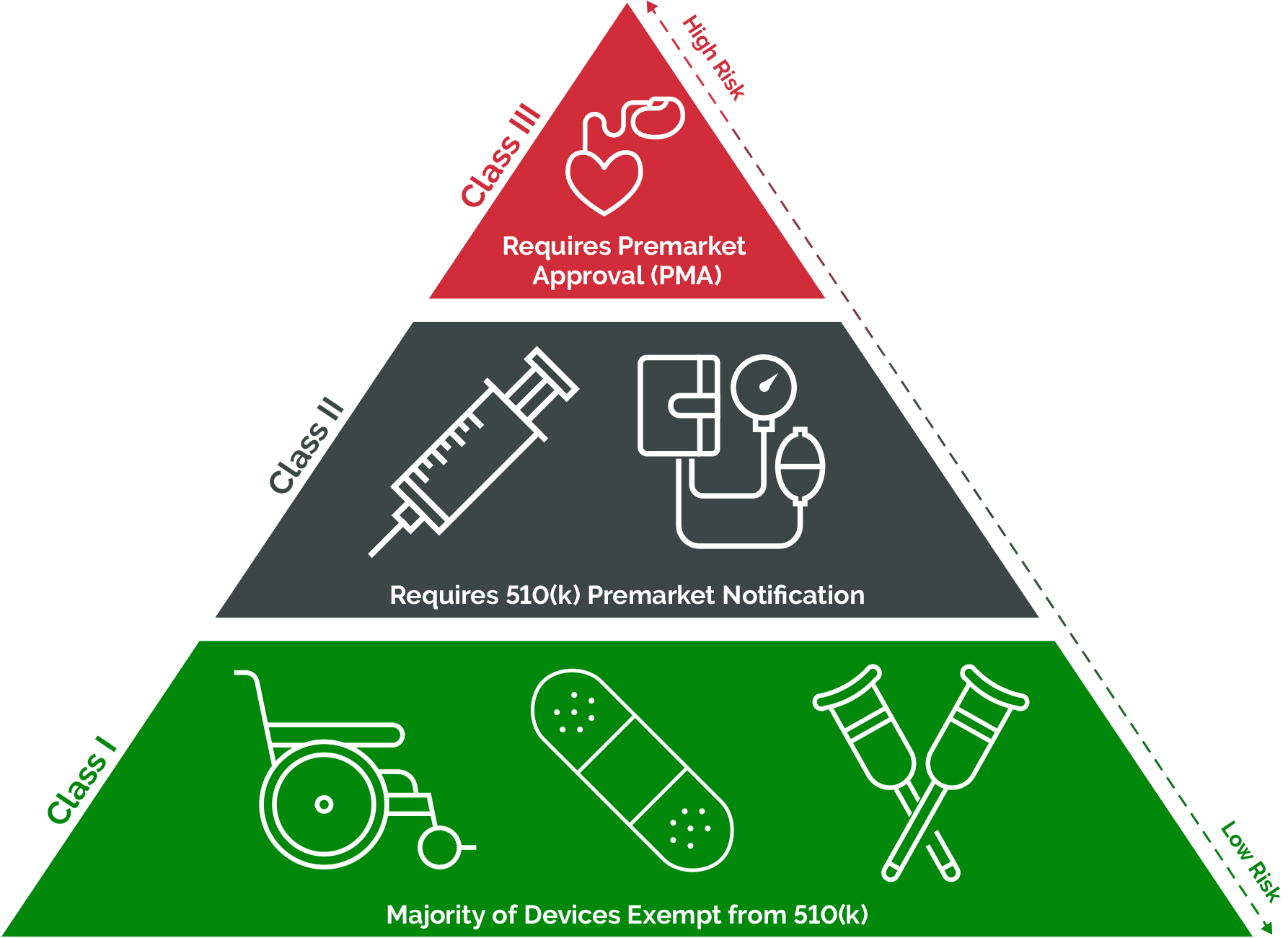
Differences between FDA medical device classes stem from the level of risk they pose and the corresponding regulatory requirements. The first step in understanding these differences is classifying devices into one of the 16 specialties outlined by the FDA. These specialties, ranging from anesthesiology to radiology, serve as organizational frameworks for over 1,700 distinct types of medical devices.
Once a device is categorized within a specialty, it falls into one of three classes: Class I, II, or III. The classification dictates the regulatory pathway and level of oversight imposed by the FDA. Class I devices, along with some exempt Class II devices, typically face fewer regulatory hurdles, often exempt from premarket notification and approval processes. Conversely, Class III and the majority of Class II devices necessitate active engagement and investigation by the FDA, including premarket notification requirements.
Additionally, some devices may qualify for exemptions based on their intended use, such as those addressing rare diseases under humanitarian device exemption. Despite these variances, all FDA-regulated devices must adhere to current Good Manufacturing Practice (cGMP) requirements concerning registration, labeling, and quality standards.
Navigating the classification system and understanding the corresponding regulatory obligations is crucial for device manufacturers to ensure compliance with FDA guidelines. This knowledge is essential in determining whether a device falls into Class I, II, or III, and whether premarket notification processes are obligatory.
FDA Medical Device Class I
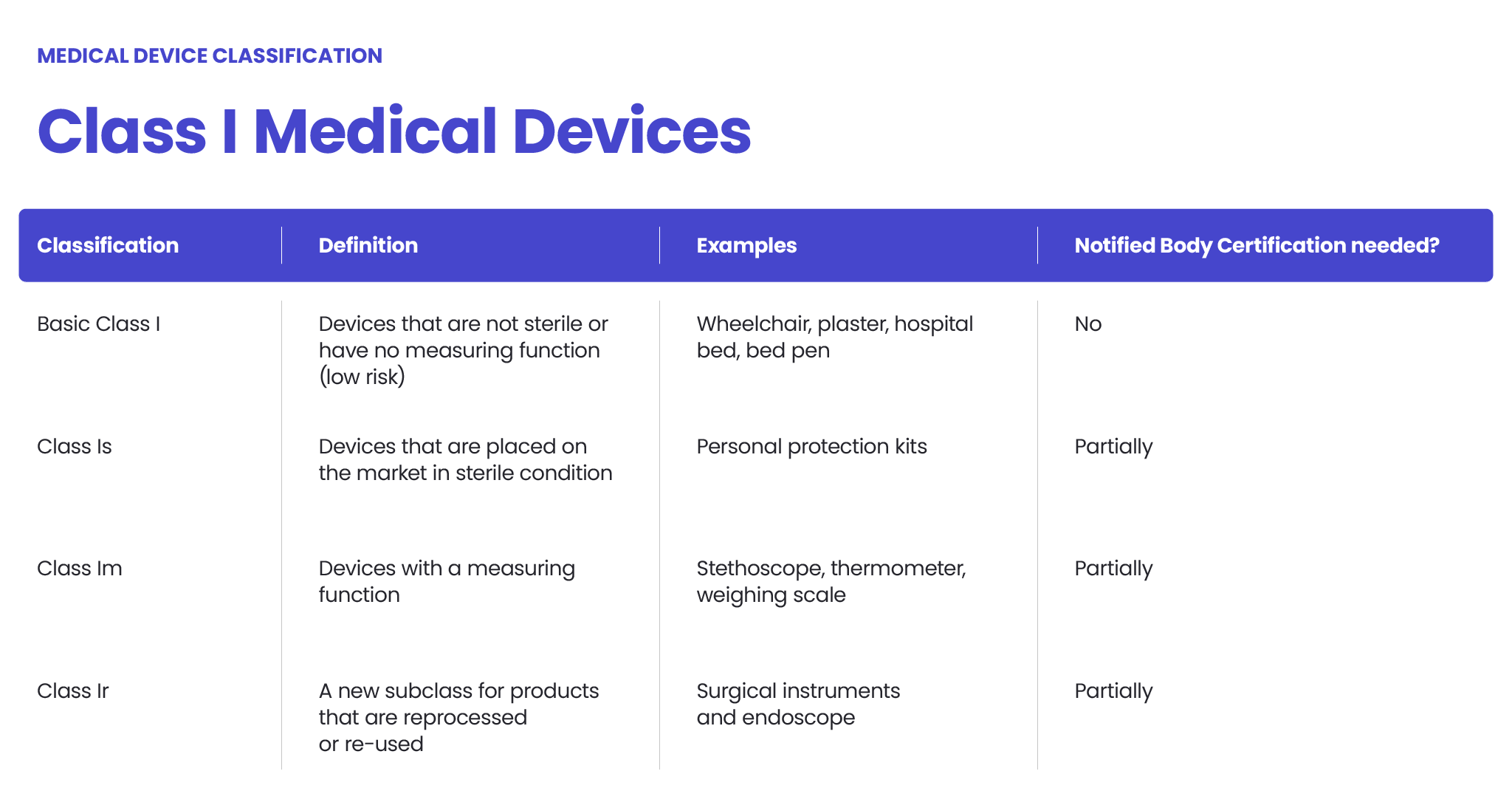
An FDA Class I medical device, as defined by the FDA, is characterized as not intended for use in supporting or sustaining life or of substantial importance in preventing impairment to human health, and they may not present a potential unreasonable risk of illness or injury. These devices represent the most common class of devices regulated by the FDA, making up 47% of approved devices on the market.
Class I devices typically have minimal contact with patients and exert low impact on overall health. Generally, they do not come into contact with a patient’s internal organs, central nervous system, or cardiovascular system. Due to their low risk nature, Class I devices are subject to the fewest regulatory requirements.
Examples of Class I medical devices include:
- Electric toothbrush
- Tongue depressor
- Oxygen mask
- Non-sterile medical gloves
- Non-electric blood pressure monitors
- Non-electric surgical instruments
- Medical neck braces
- Cervical collars
- Non-electric medical lamps
- Reusable surgical scalpel
- Bandages
- Hospital beds
- Non-electric wheelchair
Bringing Class I medical devices to market is relatively fast and straightforward compared to higher-risk devices. Since they pose the lowest risk to patients and are rarely critical to life-sustaining care, the majority of Class I devices are exempt from FDA requirements for Premarket Notification (510(k)) and Premarket Approval (PMA). However, they are not exempt from FDA General Controls, which encompass commands addressing adulteration, misbranding, device registration, records, and good manufacturing practices.
Read more: The process of registering medical devices with the FDA
FDA Medical Device Class II
Class II medical devices are characterized by a higher level of complexity and risk compared to Class I devices, primarily due to their increased likelihood of sustained contact with patients. This category encompasses devices that may interact with a patient’s cardiovascular system, internal organs, or serve diagnostic purposes.

According to the FDA, Class II devices are defined as those for which general controls alone are insufficient to ensure reasonable assurance of safety and effectiveness. Examples of Class II medical devices include:
- Catheters
- Blood pressure cuffs
- Pregnancy test kits
- Syringes
- Continuous glucose monitors
- Nebulizers
- Endoscopes
- Pulse oximeters
- Ventilators
- External defibrillators (manual)
- Dental drills
- Dental implants
- Blood transfusion kits
- Contact lenses
- Surgical gloves
- Absorbable sutures
Bringing Class II medical devices to market involves compliance with both General Controls and Special Controls. These regulations may entail device performance standards, post-market surveillance, patient registries, special labeling requirements, premarket data submission, and adherence to FDA guidelines.
A crucial aspect of the regulatory process for Class II devices is the premarket notification 510(k) process. This involves submitting a comprehensive application to the FDA, demonstrating the device’s safety and effectiveness by establishing substantial equivalence to a legally marketed predicate device. While the devices do not need to be identical, they must exhibit significant similarities in terms of use, design, materials, labeling, standards, and other characteristics
FDA Medical Device Class III
An FDA Class III medical device, as defined by the FDA, encompasses products that typically sustain or support life, are implanted, or pose a significant risk of illness or injury. This category represents a minority, constituting just 10% of devices regulated by the US FDA. Class III classification commonly applies to permanent implants, advanced medical devices, and life support systems.
Examples of Class III medical devices include:
- Breast implants
- Pacemakers
- Defibrillators
- High-frequency ventilators
- Cochlear implants
- Fetal blood sampling monitors
- Implanted prosthetics
- Implantable cardioverter-defibrillators (ICDs)
- Artificial hearts
- Ventricular assist devices (VADs)
- Deep brain stimulators
- Intracranial pressure monitors
Bringing Class III medical devices to market involves adherence to all General Controls and the FDA’s Premarket Approval (PMA) process. The FDA stipulates that general and special controls alone are insufficient to ensure the safety and effectiveness of Class III devices. Consequently, the PMA process, known for its rigor, is mandated for these devices.
While some Class III devices may qualify for the 510(k) pathway, particularly if a suitable predicate device marketed before the Medical Device Amendments of 1976 exists, this route is becoming increasingly uncommon. The FDA generally discourages the use of predicates older than a decade, making PMA the primary option for Class III devices.
The PMA process necessitates a thorough examination of the Class III medical device, requiring robust data to demonstrate safety and effectiveness. This often involves conducting clinical trials and dedicating significant time and resources to data collection. Furthermore, the FDA conducts a comprehensive review of the manufacturer’s quality system during the PMA process to ensure compliance with regulatory standards.
Summary Table Comparing Differences Between FDA Medical Device Classes:
|
Aspect |
Class I | Class II |
Class III |
| Risk Level | Lowest | Moderate | Highest |
| Examples | Tongue depressors, bandages | Blood pressure cuffs, syringes | Pacemakers, defibrillators, implants |
| Patient Contact | Minimal | Sustained | Sustained |
| Regulatory Requirements | Fewest | Moderate | Most |
| Premarket Process | Often exempt from 510(k) | Requires 510(k) clearance | Requires Premarket Approval (PMA) |
| FDA Review | General Controls only | General and Special Controls | General and Special Controls + PMA |
| Post-market Surveillance | Minimal | Moderate | Extensive |
| Risk to Patients | Lower | Moderate | Higher |
How medical devices are classified
Here’s a step-by-step guide on how to classify medical devices according to FDA regulations:
Navigate FDA Classification Regulations:
The initial step involves familiarizing yourself with the FDA classification regulations. The FDA categorizes medical devices into 16 categories based on medical specialization. These categories serve as the foundation for classifying medical devices.
Identify Relevant Category:
Let’s consider the example of classifying a blood pressure alarm. To begin, locate the appropriate category for the device. In this case, the device falls under Category 870, which pertains to cardiovascular devices.
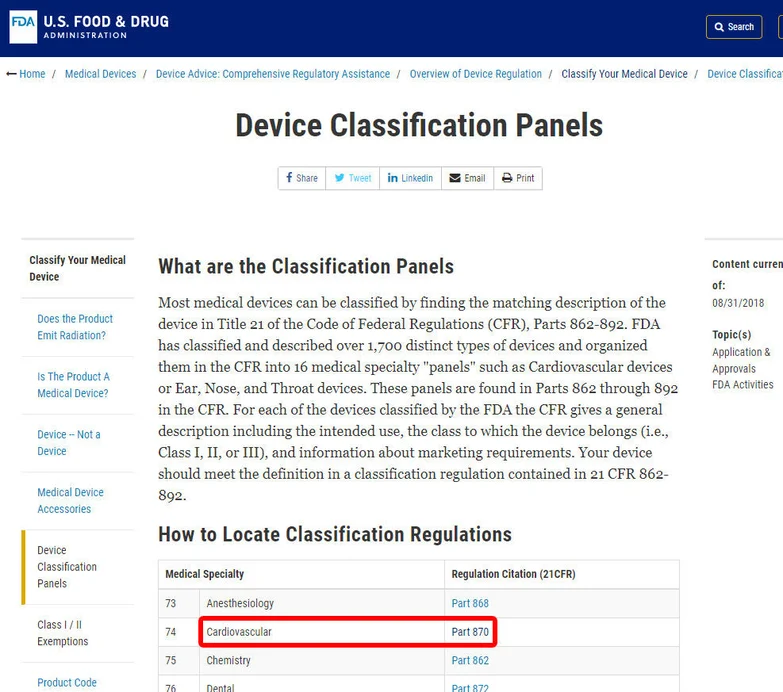
Determine Classification:
Within the identified category, search for an equivalent device and its associated device code. This step helps in pinpointing the classification of the device.
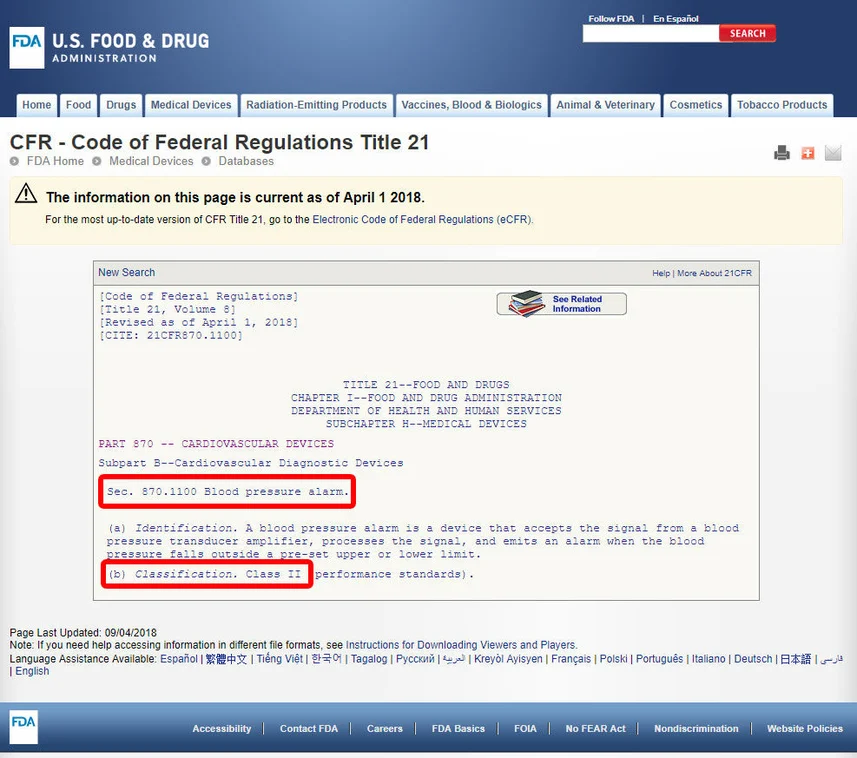
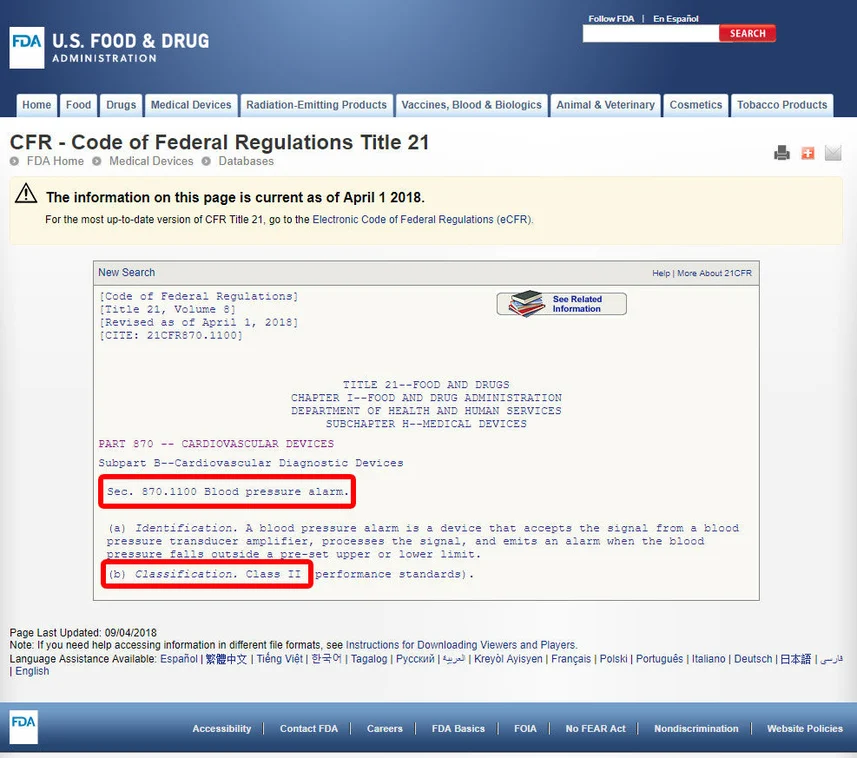
Review Guidelines:
Click on the device code to access the guidelines associated with the device. The device’s classification is typically listed under a specific section, such as section (b). For instance, upon reviewing the guidelines, we may discover that the blood pressure alarm is classified as a Class II device, indicating a medium-risk classification.
Assess for Innovation:
If your device does not have a listed equivalent among the FDA’s classified devices, it likely signifies an innovative device without a substantial equivalent. In such cases, your device would fall under Class III classification, denoting a higher level of regulatory scrutiny.
By following these steps and navigating the FDA classification regulations, you can accurately classify medical devices and determine the appropriate regulatory pathway for market approval.
Why is medical device classification important?
- Regulatory Compliance: Classification determines the level of regulatory controls and requirements imposed by regulatory authorities such as the FDA. Different classes are subject to varying degrees of scrutiny and premarket review processes to ensure safety and effectiveness.
- Risk Management: Classification reflects the level of risk associated with the device. Higher-risk devices undergo more rigorous regulatory review processes and may require additional post-market surveillance to mitigate potential risks to patients.
- Market Access: Understanding the classification helps manufacturers navigate the regulatory pathway for bringing their devices to market. It ensures compliance with regulatory requirements, expedites the approval process, and facilitates market access, thereby enabling timely availability of medical devices for patients and healthcare providers.
- Patient Safety: Proper classification ensures that medical devices are appropriately evaluated for safety and effectiveness before being introduced to the market. This safeguards patient health and reduces the likelihood of adverse events or complications associated with using medical devices.
- Innovation Support: Classification fosters innovation by providing clarity on regulatory requirements and pathways for introducing new medical technologies. It encourages manufacturers to develop novel devices while ensuring that adequate safeguards are in place to protect patient safety and public health.

Read more: What is the frequency of medical device audits by the FDA?
Rationalizing medical device classification and faster market access with GOL Solution
Streamlining the classification process and expediting market access for medical devices is paramount in today’s healthcare landscape. That’s where GOL Solution comes in, offering comprehensive FDA registration services for food facilities, medical devices, and cosmetics. Our expertise in navigating the intricate FDA regulatory framework ensures efficient and compliant registration processes, allowing manufacturers to bring their products to market faster and with confidence.
With our team of experienced professionals who possess in-depth knowledge of FDA regulations, GOL Solution offers comprehensive FDA Food registration services, FDA registration services for medical devices, and FDA registration services for cosmetics,… Contact us today for a free consultation and embark on your journey to conquer the US market!
