

The process of registering medical devices with the FDA
The process of registering medical devices with the FDA is a crucial step for companies looking to bring their products to market in the United States. With a healthcare landscape governed by stringent regulations and evolving standards, understanding and successfully navigating the FDA registration process is essential for ensuring compliance and facilitating market access. From understanding regulatory classifications to preparing comprehensive submissions and establishing robust quality management systems, the journey of medical device registration demands meticulous attention to detail and expertise. In this article, join GOL through the intricacies of the FDA registration process, explore the steps involved, and highlight key considerations for companies seeking FDA approval for medical devices their
What are the FDA medical device registration and listing requirements?
FDA medical device registration involves the process of registering establishments involved in the manufacturing, processing, packaging, labeling, importing, or distributing of medical devices with the U.S. Food and Drug Administration (FDA). This registration is a regulatory requirement for companies involved in the production and distribution of medical devices in the United States.

Below are the requirements for FDA medical device registration:
- Establishment Registration: Manufacturers, initial importers, reprocessors, and other establishments involved in medical device activities must register their facilities with the FDA. This includes both domestic and foreign establishments.
- Form FDA 2891: The registration process typically involves submitting Form FDA 2891, also known as the Establishment Registration and Device Listing form. This form collects information about the establishment, its activities related to medical devices, and the types of devices it produces or handles.
- Device Listing: In addition to establishment registration, manufacturers must also list their medical devices with the FDA. This involves submitting detailed information about each device, including its intended use, classification, and manufacturing processes, using Form FDA 2892.
- Unique Device Identifier (UDI): Most medical devices are also required to bear a Unique Device Identifier (UDI), which allows for easier identification and traceability of devices through the supply chain. Manufacturers must submit device information to the FDA’s Global Unique Device Identification Database (GUDID).
- Annual Renewal: Registrations must be renewed annually, typically between October 1 and December 31 of each year. Manufacturers must review and update their registration information as needed and submit any changes to the FDA.
- Compliance Enforcement: Compliance with FDA medical device registration requirements is essential to ensure the safety and effectiveness of medical devices marketed in the United States. Failure to register establishments or provide accurate and up-to-date information to the FDA can result in regulatory action, including fines, injunctions, or other enforcement actions.
Overall, FDA medical device registration is a crucial step in ensuring regulatory compliance and facilitating market access for medical devices in the United States. It is essential for manufacturers to understand and fulfill their registration obligations to meet FDA regulatory requirements and ensure the safety of medical devices distributed in the U.S. market.
When can I register and list a medical device with the FDA?
You can register and list a medical device with the FDA at any time, provided that you meet the necessary regulatory requirements. Here are some key points to consider:
- Establishment Registration: If you are a manufacturer, initial importer, or reprocessor of medical devices, you must register your establishment with the FDA before you begin distributing devices in the United States. This registration process involves submitting Form FDA 2891.
- Device Listing: Once your establishment is registered, you can proceed to list your medical devices with the FDA. Device listing involves providing detailed information about each device you manufacture or process, including its intended use, classification, and manufacturing processes. This information is submitted using Form FDA 2892.
- Premarket Submissions: Depending on the classification of your medical device, you may also need to submit premarket notifications (510(k)), premarket approval (PMA) applications, or de novo classification requests to the FDA before marketing your device in the United States. These submissions demonstrate the safety and effectiveness of your device and must be reviewed and approved by the FDA before you can proceed to market.
- Ongoing Compliance: Once your device is registered and listed with the FDA, you must maintain compliance with regulatory requirements, including postmarket surveillance, adverse event reporting, and updates to device listings as necessary.
It’s essential to familiarize yourself with the specific regulatory requirements applicable to your device and ensure that you meet all requirements before marketing your device in the United States. If you’re unsure about the regulatory pathway for your device or need assistance with the registration process, consulting with regulatory experts or legal counsel experienced in FDA regulations can be beneficial.
FDA medical device registration steps
The steps for FDA medical device registration involve several stages to ensure compliance with regulatory requirements and to facilitate the marketing of safe and effective medical devices in the United States. Here’s a general overview of the process:
Classification
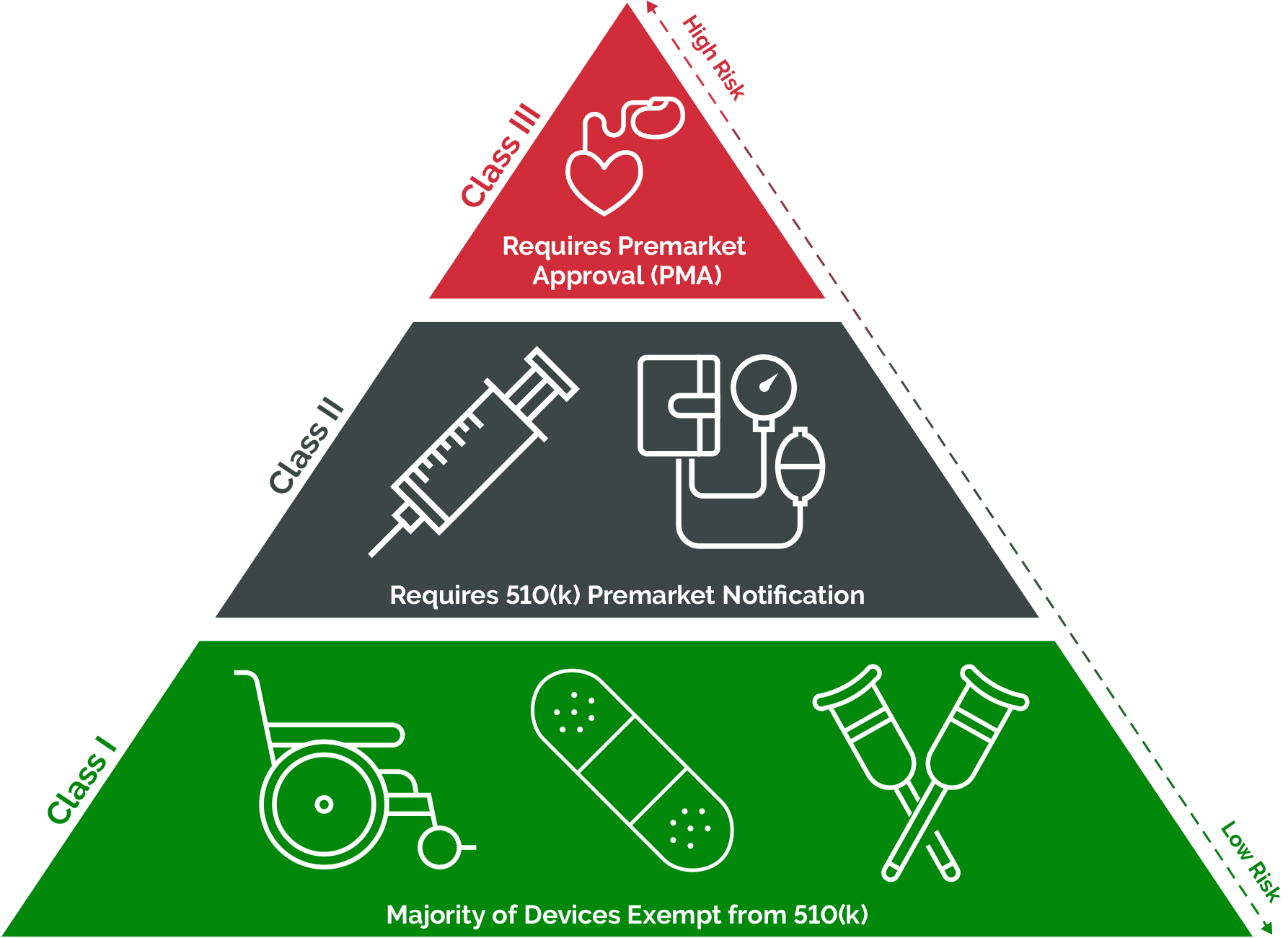
The FDA classifies medical devices into three classes based on the level of risk associated with their use:
- Class I: These devices are considered low-risk and are subject to the least regulatory controls. Examples include simple devices like bandages, tongue depressors, and handheld surgical instruments.
- Class II: Devices in this category pose moderate risk and may require special controls to ensure their safety and effectiveness. Examples include powered wheelchairs, infusion pumps, and certain diagnostic devices.
- Class III: These devices present the highest level of risk and are subject to the most stringent regulatory controls. They typically require premarket approval (PMA) to demonstrate their safety and effectiveness. Examples include implantable pacemakers, implantable defibrillators, and novel life-sustaining devices.
The FDA determines the classification of a medical device based on factors such as intended use, indications for use, technological characteristics, and the potential risks to patients. The classification process helps the FDA assign appropriate regulatory requirements to ensure the safety and effectiveness of medical devices.
The classification of a medical device determines the regulatory pathway and requirements applicable to the device:
- Class I Devices: Most Class I devices are exempt from premarket notification (510(k)) or premarket approval (PMA) requirements. However, they still need to be registered with the FDA and meet general controls, including adherence to the FDA’s Quality System Regulation (QSR).
- Class II Devices: These devices typically require a premarket notification (510(k)) demonstrating substantial equivalence to a legally marketed device (predicate device), unless they are exempt. The 510(k) process allows manufacturers to demonstrate that their device is as safe and effective as the predicate device.
- Class III Devices: Devices in this category generally require premarket approval (PMA) to demonstrate their safety and effectiveness before they can be marketed. The PMA process involves a comprehensive scientific review by the FDA, including clinical data, to evaluate the device’s safety and effectiveness.
Regardless of classification, all medical device manufacturers must comply with general controls, including establishment registration, device listing, labeling requirements, quality system regulation (QSR), and postmarket surveillance.
By understanding the classification system and associated regulatory requirements, medical device manufacturers can navigate the FDA registration and listing process effectively, ensuring compliance with regulatory standards and facilitating market access for their devices.
Registration of Establishment
Under FDA regulations, manufacturers of medical devices are required to register their establishment with the FDA. This applies to both domestic and foreign establishments that manufacture, prepare, propagate, compound, or process medical devices intended for commercial distribution in the United States.
To fulfill the registration requirement, manufacturers must submit Form FDA 2891, also known as the Establishment Registration and Device Listing form. This form collects essential information about the manufacturer’s establishment and the devices they produce.
Form FDA 2891 requires manufacturers to provide detailed information about their establishment, including:
- Name and Address: The legal name and physical address of the establishment where the medical devices are manufactured or processed.
- Contact Information: Contact details, including telephone number and email address, for communication with the FDA.
- Type of Establishment: Information about the type of establishment, such as whether it is a manufacturing facility, contract facility, or specification developer.
- Owner/Operator: The name and contact information of the owner/operator of the establishment.
- Activities Conducted: A description of the activities conducted at the establishment, including manufacturing, packaging, labeling, sterilization, and storage of medical devices.
- Device Listing: Manufacturers must also list the medical devices they produce at the establishment. This includes providing information about each device, such as its classification, intended use, and unique device identifier (UDI), if applicable.
- Submission Process: Manufacturers can submit Form FDA 2891 electronically through the FDA Unified Registration and Listing System (FURLS) or by mail. The FDA assigns a unique registration number to each establishment upon successful registration.
- Annual Renewal: Registrations must be renewed annually, typically between October 1 and December 31 of each year. Manufacturers must review and update their registration information as needed and submit any changes to the FDA.
Failure to register establishments with the FDA or providing false or misleading information on the registration form can result in regulatory action, including fines, injunctions, or other enforcement actions.
Listing of Devices
In addition to registering their establishment with the FDA, manufacturers of medical devices must also list their devices with the agency. This requirement applies to both domestic and foreign manufacturers who produce medical devices intended for commercial distribution in the United States.
To fulfill the listing requirement, manufacturers must submit Form FDA 2892, known as the Device Listing form. This form collects detailed information about each medical device manufactured or processed by the establishment.
Form FDA 2892 requires manufacturers to provide comprehensive information about each device listed, including:
- Device Identifier: A unique identifier for each device, such as the device’s name, model number, or catalog number.
- Intended Use: A description of the device’s intended use, including its indications for use and intended patient population.
- Classification: The FDA classification of the device based on its risk level (Class I, II, or III).
- Regulatory Status: Information about the device’s regulatory status, such as whether it is exempt from premarket notification (510(k)) or premarket approval (PMA) requirements.
- Manufacturing Processes: Details about the manufacturing processes involved in producing the device, including any special controls or manufacturing standards followed.
- Unique Device Identifier (UDI): If applicable, manufacturers must provide the device’s UDI, which allows for easier identification and traceability of devices through the supply chain.
- Labeling Information: Manufacturers must also provide labeling information for each device, including labeling claims, warnings, instructions for use, and any applicable symbols or markings.
- Submission Process: Similar to establishment registration, manufacturers can submit Form FDA 2892 electronically through the FDA Unified Registration and Listing System (FURLS) or by mail. Each listed device is assigned a unique identifier by the FDA.
- Annual Updates: Manufacturers are required to update their device listings annually, as well as whenever there are changes to device information, such as changes in manufacturing processes, labeling, or intended use. Updates must be submitted to the FDA within 30 days of any changes.
Failure to list devices with the FDA or providing false or misleading information on the listing form can result in regulatory action, including fines, injunctions, or other enforcement actions.
Quality System Regulation (QSR)
The FDA’s Quality System Regulation (QSR) is outlined in 21 CFR Part 820 of the Code of Federal Regulations. It establishes comprehensive requirements for the design, manufacture, packaging, labeling, storage, installation, and servicing of medical devices marketed in the United States.

QSR applies to most medical devices, regardless of their classification or regulatory pathway. It encompasses a wide range of activities involved in the lifecycle of medical devices, from initial design and development to postmarket surveillance and servicing.
Some of the key requirements outlined in the FDA’s Quality System Regulation include:
- Management Responsibility: Manufacturers are required to establish and maintain a quality management system (QMS) that ensures compliance with regulatory requirements and promotes the safety and effectiveness of medical devices.
- Design Controls: Manufacturers must establish and maintain procedures for design controls to ensure that medical devices meet specified design requirements and intended uses. This includes conducting design reviews, verifying and validating designs, and documenting design changes.
- Document Controls: Manufacturers must establish and maintain procedures for document control to ensure that documents related to the QMS are adequately controlled, reviewed, and approved.
- Production and Process Controls: Manufacturers must establish and maintain procedures for production and process controls to ensure that medical devices are manufactured, packaged, labeled, and stored in accordance with specified requirements.
- Labeling and Packaging Controls: Manufacturers must establish and maintain procedures for labeling and packaging controls to ensure that medical devices are properly labeled and packaged for their intended use.
- Device History Records (DHR): Manufacturers must maintain device history records (DHR) for each batch, lot, or unit of medical device manufactured. These records must include information about the manufacturing process, including dates, quantities, and personnel involved.
- Corrective and Preventive Actions (CAPA): Manufacturers must establish and maintain procedures for corrective and preventive actions (CAPA) to address nonconformities and prevent recurrence of quality issues.
- Complaint Handling: Manufacturers must establish and maintain procedures for handling complaints related to medical devices, including investigation, evaluation, and documentation of complaints.
- Compliance and Enforcement: Compliance with the FDA’s Quality System Regulation is subject to inspection by the FDA and other regulatory authorities. Non-compliance can result in regulatory action, including warning letters, fines, injunctions, or other enforcement actions.
Unique Device Identifier (UDI)
The Unique Device Identifier (UDI) system is a regulatory requirement established by the FDA to enhance the identification and traceability of medical devices throughout the supply chain. UDI implementation is mandated for most medical devices marketed in the United States.
The primary purpose of the UDI system is to improve patient safety by facilitating more accurate and efficient identification, tracking, and tracing of medical devices. By assigning a unique identifier to each device, stakeholders can more easily locate and recall devices in the event of safety issues or adverse events.
Components of UDI: The UDI consists of two main components:
- Device Identifier (DI): A unique numeric or alphanumeric code assigned to a specific version or model of a device. The DI identifies the specific device labeller and the specific version or model of the device.
- Production Identifier (PI): Additional information that identifies specific units of the device, such as the lot number, serial number, expiration date, and manufacturing date.
Manufacturers are required to submit UDI information to the FDA’s Global Unique Device Identification Database (GUDID). The GUDID serves as a publicly accessible repository of device identification information, allowing stakeholders to search for and retrieve information about specific medical devices.
- Information Submitted to GUDID: Manufacturers must submit detailed information about each medical device, including:
- Device Identifier (DI)
- Product Identifier (PI)
- Device Description
- Model Number
- Brand Name
- Regulatory Information (e.g., FDA product code, class, submission type)
- Manufacturing Information (e.g., manufacturer name, location)
- Labeler Information (e.g., labeler name, contact information)
- Package Information (e.g., package quantity, type)
- Date Information (e.g., manufacturing date, expiration date)
Compliance Deadlines: The FDA has established deadlines for compliance with UDI requirements based on device classification and risk. Class III devices and devices licensed under the Public Health Service Act (PHS Act) were required to comply with UDI requirements earlier, while Class II devices have staggered compliance deadlines.
Benefits of UDI: Implementation of the UDI system offers several benefits, including:
- Improved patient safety through enhanced device identification and traceability
- More efficient and accurate adverse event reporting and device recalls
- Enhanced supply chain management and inventory control
- Facilitated postmarket surveillance and research
Premarket Notification (510(k)), Premarket Approval (PMA), or De Novo Classification
Premarket Notification (510(k)):

Manufacturers of Class II medical devices may be required to submit a premarket notification (510(k)) to the FDA.
The purpose of the 510(k) submission is to demonstrate that the new device is substantially equivalent to a legally marketed predicate device with respect to intended use, technological characteristics, and performance.
Submission Process: The manufacturer prepares and submits a 510(k) application to the FDA, along with supporting documentation and data comparing the new device to the predicate device.
Review Process: The FDA evaluates the 510(k) submission to determine whether the new device is substantially equivalent to the predicate device. If deemed substantially equivalent, the new device can proceed to market.
Premarket Approval (PMA):
Manufacturers of Class III medical devices are typically required to submit a premarket approval (PMA) application to the FDA.
The purpose of the PMA application is to demonstrate the safety and effectiveness of the new device through comprehensive scientific evidence, including clinical data.
Submission Process: The manufacturer prepares and submits a PMA application to the FDA, including detailed information about the device, preclinical and clinical data, manufacturing processes, and labeling.
Review Process: The FDA conducts a thorough review of the PMA application, including evaluation of preclinical and clinical data, manufacturing processes, and labeling. Approval of the PMA application indicates that the FDA has determined the device to be safe and effective for its intended use.

De Novo Classification
Manufacturers of novel devices that do not have a legally marketed predicate device may submit a de novo classification request to the FDA.
The purpose of the de novo classification request is to establish a new regulatory classification for the device based on its risk and intended use.
Submission Process: The manufacturer prepares and submits a de novo classification request to the FDA, providing detailed information about the device, its intended use, technological characteristics, and any available data supporting safety and effectiveness.
Review Process: The FDA evaluates the de novo classification request to determine whether the device meets the criteria for a new regulatory classification. If granted, the device can proceed to market under the newly established classification.

Labeling Requirements
The FDA regulates medical device labeling under 21 CFR Part 801, which outlines requirements for the labeling of medical devices distributed in the United States.
Medical device labeling must be clear, accurate, and informative to ensure safe and effective use of the device. It should provide essential information about the device, its intended use, and any necessary precautions or warnings.
The labeling of medical devices typically includes the following elements:
- Device Identification: The label must include the name and intended use of the device, as well as the manufacturer’s name and address.
- Instructions for Use: Clear and concise instructions for using the device safely and effectively must be provided. This includes information on device preparation, operation, maintenance, and disposal.
- Warnings and Precautions: Labels must prominently display any warnings or precautions associated with the use of the device, including potential risks or adverse effects.
- Indications for Use: The label should specify the intended use of the device and any limitations or contraindications.
- Storage and Handling Instructions: If applicable, labels should include recommendations for storing and handling the device to maintain its safety and effectiveness.
- Symbol and Markings: Labels may include standardized symbols or markings as specified by FDA regulations. These symbols convey important information, such as the presence of radiation, sterilization requirements, or environmental considerations.
- Format and Layout: FDA regulations specify requirements for the format and layout of medical device labeling to ensure readability and comprehension. Labels should use clear and legible fonts, appropriate sizing, and sufficient contrast between text and background.
- Multilingual Labeling: For devices distributed internationally or intended for use by non-English-speaking populations within the United States, manufacturers may need to provide labeling in multiple languages to ensure comprehension.
Manufacturers are responsible for ensuring that device labeling complies with FDA regulations before distribution. Labeling content may be subject to FDA review as part of the premarket submission process or during routine inspections.
Manufacturers must maintain up-to-date labeling for their devices and promptly update labels to reflect any changes in device design, indications for use, warnings, or other relevant information.
Postmarket Surveillance
Postmarket surveillance is a critical component of the FDA’s regulatory oversight of medical devices, aimed at monitoring device performance and identifying potential safety issues or adverse events that may arise after devices are marketed.
Manufacturers are responsible for establishing procedures for postmarket surveillance of their devices to monitor and evaluate their safety and effectiveness in real-world use.
Manufacturers are required to promptly report adverse events associated with their devices to the FDA. Adverse events include any serious injuries, deaths, or malfunctions that may occur as a result of device use.
Types of Adverse Events:
- Serious Injuries: Any injury that results in death, serious injury, or requires medical intervention to prevent permanent impairment or damage.
- Deaths: Any death that occurs as a result of device use, regardless of whether the device directly caused the death.
- Malfunctions: Any failure or malfunction of the device that could directly cause or contribute to a serious injury or death if the malfunction were to recur.
Manufacturers must submit adverse event reports to the FDA in accordance with regulatory requirements. This typically involves completing and submitting Form FDA 3500A, also known as the MedWatch form, electronically or by mail.
Manufacturers must report adverse events to the FDA in a timely manner. The specific reporting timeframe may vary depending on the severity of the event, but generally, serious injuries, deaths, and malfunctions must be reported within a specified timeframe after the manufacturer becomes aware of the event.
Manufacturers are also responsible for investigating reported adverse events to determine the root cause and implementing any necessary corrective or preventive actions to mitigate the risk of recurrence.
Manufacturers may develop postmarket surveillance plans to systematically monitor device performance, identify trends or patterns of adverse events, and assess the need for additional safety measures or device modifications.
The FDA monitors and evaluates adverse event reports submitted by manufacturers to identify potential safety issues or emerging trends. The agency may take regulatory action, such as issuing safety communications, requiring labeling changes, or ordering device recalls, based on the findings of postmarket surveillance activities.
Device Listing Updates
Manufacturers are required to review and update their device listings annually to ensure that the information provided to the FDA remains accurate and up-to-date. These annual updates help maintain the integrity of the FDA’s device listing database and ensure that stakeholders have access to current information about medical devices on the market.
Manufacturers must also update their device listings whenever there are changes to device information that may affect its regulatory status or safety. Examples of changes that may require updates include:
- Changes in Manufacturing Location: If the manufacturing location of the device changes, manufacturers must update their device listings to reflect the new location.
- Modifications to the Device: Any modifications to the design, materials, manufacturing processes, or labeling of the device that could affect its safety, effectiveness, or regulatory status must be reported to the FDA and reflected in the device listing.
- Changes in Ownership or Contact Information: Manufacturers must update their device listings to reflect changes in ownership, contact information, or other relevant details about the establishment responsible for the device.
Manufacturers can update their device listings through the FDA’s Unified Registration and Listing System (FURLS) or by submitting a revised Form FDA 2892. The updated information should be submitted to the FDA within 30 days of any changes.
Failure to update device listings in a timely manner or providing inaccurate or incomplete information to the FDA can result in regulatory action, including fines, injunctions, or other enforcement actions.
Manufacturers should establish procedures for monitoring changes to device information and ensure that updates to device listings are made promptly and accurately. This may involve implementing internal processes for tracking changes, conducting periodic reviews of device listings, and assigning responsibility for submitting updates to designated personnel.
Tips for medical device registration

Navigating the FDA medical device registration process can be complex, but here are some tips to help you successfully register your medical device:
Understand Regulatory Requirements
Familiarize yourself with FDA regulations and guidance documents relevant to medical device registration, including classification, premarket submission requirements, quality system regulation (QSR), labeling requirements, and postmarket surveillance.
Plan Early
Start planning for FDA registration early in the development process of your medical device. Understanding regulatory requirements and incorporating them into your development plan can help streamline the registration process later on.
Seek Expert Guidance
Consider consulting with regulatory experts or hiring regulatory consultants who specialize in medical device registration. They can provide valuable insights and guidance to navigate the regulatory requirements effectively.
Establish a Quality Management System
Implement a robust quality management system (QMS) that complies with the FDA’s Quality System Regulation (QSR) outlined in 21 CFR Part 820. Having a well-documented QMS in place demonstrates your commitment to producing safe and effective medical devices.
Thoroughly Prepare Premarket Submissions
If your device requires premarket notification (510(k)), premarket approval (PMA), or De Novo classification, ensure that you thoroughly prepare the submission documentation. This includes compiling comprehensive scientific evidence, comparative testing data, and other relevant information to support the safety and effectiveness of your device.
Ensure Accurate Device Listing
Provide accurate and detailed information when listing your medical devices with the FDA. Double-check the information submitted on Form FDA 2892 to avoid errors or discrepancies that could delay the registration process.
Stay Up-to-Date
Stay informed about any updates or changes to FDA regulations, guidance documents, and submission requirements. Regularly check the FDA’s website for updates and subscribe to FDA email alerts to receive timely notifications.
Communicate with the FDA
Maintain open communication with the FDA throughout the registration process. If you have any questions or need clarification on regulatory requirements, don’t hesitate to reach out to the FDA for guidance.
Prepare for Postmarket Requirements
Be prepared to comply with postmarket surveillance requirements, including monitoring and reporting adverse events associated with your device. Establish procedures for postmarket surveillance and ensure timely reporting to the FDA as required.
Seek Continuous Improvement
Continuously evaluate and improve your regulatory compliance processes to ensure ongoing compliance with FDA regulations. Regularly assess your QMS, labeling, and postmarket surveillance procedures to identify areas for improvement.
Do all medical devices require FDA approval?
Not all medical devices require FDA approval before they can be marketed in the United States. The level of regulatory control imposed by the FDA depends on the risk associated with the device. The FDA classifies medical devices into three main categories based on their risk level: Class I, Class II, and Class III.
- Class I Devices: These are considered low-risk devices, such as tongue depressors, bandages, and most non-invasive diagnostic devices. Most Class I devices are exempt from the FDA’s premarket notification (510(k)) or premarket approval (PMA) requirements. However, they still need to be registered with the FDA and meet general controls, including adherence to the FDA’s Quality System Regulation (QSR).
- Class II Devices: These are moderate-risk devices, such as infusion pumps, surgical gloves, and some diagnostic imaging devices. Class II devices generally require a premarket notification (510(k)) demonstrating substantial equivalence to a legally marketed device (predicate device) unless they are exempt. The 510(k) process allows manufacturers to demonstrate that their device is as safe and effective as the predicate device.
- Class III Devices: These are high-risk devices, such as implantable pacemakers, implantable defibrillators, and some life-sustaining devices. Class III devices typically require premarket approval (PMA) to demonstrate their safety and effectiveness before they can be marketed. The PMA process involves a comprehensive scientific review by the FDA to evaluate the device’s safety and effectiveness based on clinical data.
In addition to premarket requirements, all medical device manufacturers must register their establishments with the FDA and list their devices with the agency. They must also comply with general controls, including adherence to the FDA’s Quality System Regulation (QSR), labeling requirements, and postmarket surveillance.
The FDA’s regulatory oversight aims to ensure that medical devices marketed in the United States are safe, effective, and reliable for their intended use. The level of regulatory scrutiny reflects the potential risks associated with the device and helps to safeguard public health and safety.
FDA medical device registration assistance with GOL
FDA medical device registration assistance with GOL offers comprehensive support and expertise in navigating the complex regulatory landscape of FDA regulations. Below are some extremely outstanding capabilities of GOL:
- Logistics Expertise: GOL brings over 25 years of experience in the supply chain industry, specializing in logistics solutions tailored to the unique requirements of medical device registration with the FDA.
- Information Technology Specialist: With a pioneering background in developing technology solutions for the logistics industry since 2002, GOL leverages cutting-edge IT solutions to streamline the FDA registration process for medical devices.
- Regulatory Compliance: GOL is a trusted expert in e-logistics, e-compliance, and e-government solutions, ensuring that clients meet all regulatory requirements set forth by the FDA for medical device registration.
- Solution Builder: GOL is a leading solution builder for supply chain compliance and optimization solutions, offering tailored assistance to clients seeking FDA medical device registration.
- Values: GOL upholds essential values of sustainability, efficiency, and trust, ensuring that clients and global partners receive reliable and high-quality services throughout the FDA registration process.
With GOL’s expertise and commitment to excellence, clients can navigate the FDA medical device registration process with confidence, knowing that they have a trusted partner by their side every step of the way.
In conclusion, registering medical devices with the FDA is a critical process that requires careful attention to detail and adherence to regulatory standards. With the assistance of GOL, a trusted expert in logistics and regulatory compliance, manufacturers can navigate this complex journey with confidence. By leveraging GOL’s expertise, companies can streamline the FDA registration process, ensure compliance, and ultimately bring their medical devices to market efficiently and effectively.
